


📢 转载信息
原文链接:https://www.nature.com/articles/s41586-025-10093-z
原文作者:Lucía Barbadilla-Martínez, Noud Klaassen, et al. (Nature)
摘要
启动子是所有基因的核心调控元件。它们的活性确保了每个基因的正确转录水平,这对细胞稳态和对各种信号的反应至关重要。基因组学中的一个主要挑战是构建能够从调控元件序列中准确预测全基因组基因表达的计算模型1。在此,我们介绍了启动子活性调控模型 (PARM),这是一个细胞类型特异性的深度学习模型,使用专门设计的全并行报告基因分析 (MPRAs) 进行训练,该分析用于查询人类启动子序列。PARM 在实验上和计算上都非常轻量,因此可以生成细胞类型特异性和条件特异性的模型,这些模型可以仅从DNA序列可靠地预测全基因组范围内的自主启动子活性。PARM 还可以设计纯合成的强启动子。我们利用 PARM 系统地识别了可能有助于每个天然人类启动子活性的转录因子 (TF) 结合位点,并检测了细胞暴露于各种刺激后这些调控相互作用的重新布线情况。我们还揭示并实验确认了转录因子在激活性和抑制性调控功能之间存在实质性的位置偏好,以及基序-基序相互作用的复杂语法。我们的方法提供了一种高度经济的策略,以更深入地理解转录因子对人类启动子的动态调控。
正文
启动子通常由一个转录起始位点 (TSS) 和几百个碱基对的DNA(大多位于TSS上游)组成,其中包含可以被多种转录因子 (TFs) 结合的短序列基序2,3。构建能够从DNA序列预测启动子活性的计算模型是具有挑战性的。深度学习技术为此目的带来了希望1,4,5,6,但依赖于大量的训练数据集。在一种方法中,这种数据量是通过汇总来自多种细胞类型的数百或数千个基因转录和表观遗传特征的全基因组图谱获得的7,8,9,10,11,12,13。训练此类模型需要大量的计算能力。此外,表观遗传学特征是调控元件活性的不完美的替代指标14,可能受到长程自相关模式的混淆15,并且只是相关的。因此,从这种图谱数据中推断DNA序列与启动子活性之间的因果联系在本质上是困难的。尽管如此,一些由此产生的模型已被用于揭示人类启动子调控语法的特性,例如转录起始信号、双向转录的序列基础12和TF-TF相互作用13。然而,这些深度学习模型无法预测原始大型训练集中不包含的细胞类型或条件下的基因调控变化。
MPRAs提供了一种替代的训练数据来源,可以针对单个特定细胞类型获得。在MPRA中,数百万个基因组DNA序列(每个长度几百个碱基对)在目标细胞类型中测试其自主调控活性。由于片段是隔离测试的,测得的活性可以明确分配;因此,推断特定DNA序列的因果作用可能比使用表观遗传图谱和转录组图谱更容易(图 1a)。在早期探索16之后,已有报道表明MPRA与深度学习相结合可以为果蝇和人类增强子17,18,19,20以及酵母启动子21产生强大的预测模型。然而,在人类启动子上的应用18,19迄今为止仅限于少数几种细胞类型,并且其调控语法的许多方面仍未被探索。
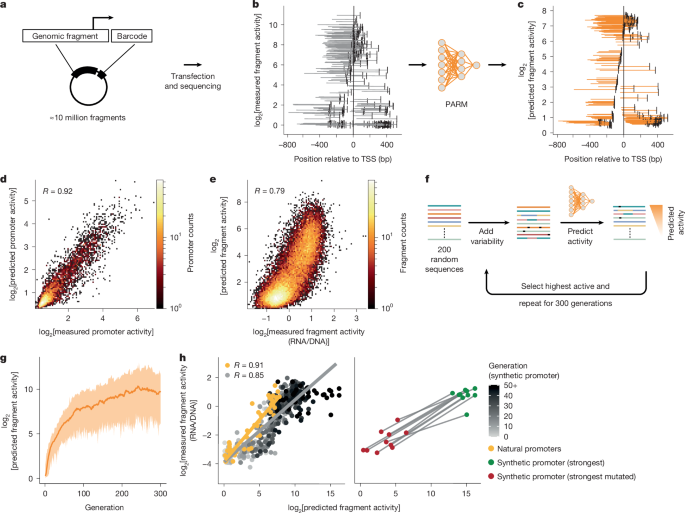
a, 通过条形码 MPRA22 测量数百万个随机 gDNA 片段自主启动子活性的示意图。b, 一个典型的 MPRA 结果,显示了与VCPKMT启动子重叠的片段。数据来自 K562 细胞22。水平线,右侧有垂直刻度,显示片段位置和测得的启动子活性(方法)。c, PARM 对 b 中相同片段的预测活性。d, 从训练和验证数据中随机剔除的 5,204 个有趣启动子的预测活性与实测活性之间的相关性。启动子活性定义为所有重叠片段的平均活性。MPRA 数据来自参考文献22,PARM 预测显示的是 K562 细胞中的情况。e, 使用慢病毒介导的 MPRA 在 K562 细胞中对启动子片段的预测活性与实测活性之间的相关性19。f, 用于迭代生成高活性合成启动子的遗传算法工作流程,改编自先前研究21。从 200 个随机序列(各 232 bp)开始,300 次迭代中的每一次都包括随机诱变和重组,然后选择 PARM 预测的最强启动子。我们在十个独立实例中运行了遗传算法。g, 一个遗传算法实例中预测的启动子活性的增加。橙色曲线,群体的平均预测启动子活性;阴影,活性的全范围。h, K562 细胞中 MPRA 对 232 bp 合成启动子活性的实验验证。左侧,PARM 预测的活性和测得的 42 个天然人类启动子片段(橙色)和 392 个从不同代数选择的合成启动子(灰度)的预测活性。右侧,最强的 10 个合成启动子(绿色)(每个遗传算法实例中一个)以及突变了 12–18 个关键核苷酸后的相同启动子(红色)。R 值表示皮尔逊相关系数。
在此,我们提出了一个平台,将优化的 MPRA 与深度学习相结合,以高效地构建所有人类启动子的序列-活性模型。通过这个名为 PARM 的平台,数据生成和计算建模都非常经济。这一发展使我们能够在十种不同的细胞类型中,以及在细胞暴露于多种刺激后,构建序列-活性模型。
深度学习模型预测启动子活性
我们首先通过使用 MPRA 的数据进行训练,构建了一个基于序列的深度学习启动子活性模型,该 MPRA 测量了数百万个 gDNA 片段的自主启动子活性。这些数据是在人 K562 骨髓性白血病细胞和 HepG2 肝细胞癌细胞中生成的22。至关重要的是,基因组中每个位置平均被大约 240 个随机、部分重叠的片段覆盖(图 1b)。为了训练深度学习模型,我们总共选择了大约 1000 万个 88–600 bp 的片段,它们与 30,607 个经过筛选的人类启动子重叠。PARM 的架构是一个卷积神经网络 (CNN),先前已用于模拟类似的数据结构16,17,21,23。我们广泛优化了 CNN 架构和超参数(扩展数据图 1a,b)。除了 MPRA 数据外,训练过程中未引入任何先验知识。我们为每种细胞系训练了一个单独的模型。
交叉验证显示,PARM 能够以很高的准确性预测随机剔除的 5,204 个启动子的实测活性(K562 细胞中皮尔逊R = 0.92,HepG2 细胞中 R = 0.89;图 1d 和扩展数据图 1c)。在给定启动子中,PARM 也准确地预测了单个被测片段的活性(图 1b,c)。此外,PARM 在 K562 和 HepG2 细胞中,使用附着体和慢病毒介导的 MPRA19,24 测试的启动子片段的活性具有可靠的预测能力(R = 0.78–0.80)(图 1e 和扩展数据图 1d–f)。这些发现表明,在附着体 MPRA 上训练的 PARM 可以准确估计整合到基因组中的启动子的活性25。
这些 PARM 模型使我们能够进行从内到外的饱和突变分析 (ISM),以预测单个核苷酸取代对每个启动子活性的影响7,10,26。当应用于TERT癌基因的启动子时,PARM 正确预测了大约 25% 的人类癌症中发生的 C250T 和 C228T 突变会导致TERT表达增加27(扩展数据图 2a,黑点)。它还预测了在人类肿瘤中低频率发生的其他几个TERT启动子突变的活性增加(扩展数据图 2b),并正确识别了TERT启动子活性所必需的几个位置28(扩展数据图 2a,黑条)。使用 K562 细胞数据训练的 PARM 还可以预测血液中顺式作用表达数量性状位点 (eQTLs) 的效应29。与使用数千个表观遗传数据集训练的两个最先进的深度学习模型(Enformer10 和 Borzoi30)相比,PARM 对这些顺式作用 eQTL 的预测准确性与 Enformer10 相似,但不如 Borzoi30(扩展数据图 2c,d)。然而,Borzoi 的计算要求更高,因为它包含超过 3000 万个拟合参数,而 PARM 只有 742,337 个。
设计完全合成的启动子
为了进一步测试 PARM,我们研究了它是否可以自动生成合成的人类启动子。为此,我们改编了一个遗传算法21,该算法迭代地诱变一组序列,并选择那些具有预测最高启动子活性的序列(图 1f)。从随机序列开始,这个过程不断产生一系列序列,PARM 预测它们是高活性启动子(图 1g)。我们使用 MPRA 对 455 个此类合成序列进行了实验测试,这些序列具有广泛的预测活性(扩展数据图 3a,b),以及 42 个天然人类启动子序列,包括 K562 细胞中活性最高的 15 个天然人类启动子(补充数据 1)。正如预期的那样,天然启动子片段的实测活性与预测活性高度相关(图 1h)。合成启动子也是如此,尽管在最高的预测活性时观察到了饱和现象(图 1h)。在测量中,预测活性最高的合成启动子与最强的天然启动子活性大致相同。该模型准确地标定出了影响启动子活性的序列元件。也就是说,对 10 个活性最高的合成片段中预测最关键的 12–18 个核苷酸进行突变,导致实测活性平均降低 3.16 ± 0.77 倍(平均值 ± 标准差)(图 1h)。尽管合成序列是基于 K562 细胞的数据使用 PARM 模型优化的,但当在 HepG2 细胞中测试相同文库的 232 bp 片段时,我们在实测活性和预测活性之间得到了相似的相关性(扩展数据图 3c)。当我们生成仅 70 bp 的合成启动子时,我们得到了相似的结果,尽管预测活性和实测活性具有较小的动态范围(扩展数据图 3c)。这一发现表明,强启动子通常必须长于 70 bp。
合成启动子与人类基因组中的任何序列都没有显著相似性(使用 BLAST31 搜索评估),这证明 PARM 并非简单地复制现有的人类启动子序列。显示出强活性的合成启动子通常在 K562 细胞中具有已知激活因子 TF 家族(如 FOS–JUN、ETS 和 CREB 相关因子)的基序。然而,每个启动子携带不同的基序组合(扩展数据图 3d)。因此,PARM 可以通过组合它已经学到在测试细胞类型中活跃的 TF 结合基序,从头开始生成高活性启动子。这一结果表明 PARM 已经学习了人类启动子调控语法的生物学相关方面。
功能性 TF 基序的推断
PARM 的计算效率使我们能够应用 ISM 来分析 30,607 个人类启动子(可在在线交互式浏览器 http://parm.deridderlab.nl 中查看)。对于 K562 细胞,该分析通常识别出一个或多个 4–10 个相邻核苷酸的区域,这些区域被一致地预测为增强或降低活性(扩展数据图 4a)。为了识别假定的相关 TF,我们将这些模式与所有 TF 的已知结合特异性32进行匹配,对于几乎所有 TF,估计的错误发现率 (FDR) < 5%(补充图 1)。如下例所示,这些结果图谱不仅仅描述了基于 DNA 序列的假定 TF 结合位点,还预测了影响启动子活性的功能性结合位点(此后称为调控位点,RSs)。
与 RSs 相关的多数 TF 确实如 mRNA 测序数据显示的那样在 K562 细胞中表达。对于未表达的 TF,其同源基序与表达的 TF 高度相似。当考虑到这一点后,几乎所有检测到的基序都可以与表达的 TF 相关联(扩展数据图 4b)。在 K562 细胞中,这导致 20,543 个具有至少一个 RS 的启动子(扩展数据图 4c)。值得注意的是,许多 RSs 与具有高度相似结合特异性的多个 TF 相匹配,因此被注释了不止一次。不出所料,剩余的 10,064 个没有任何预测 RS 的启动子,在 MPRA 和内源性表达中通常活性要低得多(扩展数据图 4d)。
接下来,我们想知道人类启动子是否可能具有不对应于任何已知 TF 结合基序的额外 RSs。我们扫描了所有启动子中至少有五个相邻核苷酸的区域,这些区域被一致地预测为增强或降低启动子活性。在 K562 细胞中,此分析产生了 38,434 个与至少 1 个已知 TF 基序匹配的 RSs,以及 1,402 个不匹配任何已知基序的 RSs(扩展数据图 5a)。因此,即使 PARM 从未提供 TF 基序信息,它也强烈地收敛于已知的 TF 生物学。在罕见的未注释的 RS 序列中,我们确定了 10 个簇,每个簇在人类启动子中至少包含 30 个相似的 RSs(扩展数据图 5a)。
簇 3 在人类启动子中出现 57 次,其共有序列为 TCTCTATGGT。为了确定结合该基序的假定 TF,我们使用 K562 细胞的核提取物和包含该基序的启动子序列进行了 DNA 亲和纯化(扩展数据图 5b),随后进行基于定量质谱的蛋白质组学分析33。该分析显示,ZNF48 是最可能的结合该基序的 TF。先前基于染色质免疫沉淀数据34,已为该 TF 推断出 TCTCT 结合基序。使用昆虫细胞中产生的核提取物和重组 ZNF48 进行的附加体外结合实验证实了 ZNF48 与簇 3 基序的直接结合(扩展数据图 5c–f 和补充图 2a–f)。簇 5 的基序最近被发现由 ZFP9135 结合。因此,PARM 可以识别功能性 TF 基序,这些基序相对不常见且注释不佳,但可能具有先前未知的调控相互作用。
以启动子为中心的 MPRA 文库
如上所述,使用高复杂性文库的 MPRA 实验在技术上具有挑战性,因为它们需要大量细胞和高转染效率22。这一要求大大限制了可扩展性。因为我们仅使用来自启动子重叠片段的数据来训练 PARM,我们推断一个仅包含此类片段的 MPRA 文库就足够了。因此,我们使用一种捕获策略创建了新的 MPRA 文库,这些文库高度富集(90%)了来自人类基因组文库的启动子重叠片段(图 2a 和扩展数据图 11a–e)。一个文库包含 400 万个充分代表的唯一片段,比组合的全基因组文库22少约 600 倍。然而,它仍然平均覆盖了所有人类 TSS 约 151 倍(扩展数据图 11c),并且片段大小具有广泛的多样性(扩展数据图 11a)。该文库用于下面描述的大多数实验(第二个文库有 1100 万个片段,用于少数实验,并且获得了相似的结果;扩展数据图 11a–d)。这些聚焦的文库使我们能够在两次独立的重复实验中仅使用大约 500 万个细胞;即,比全基因组 MPRAs 22 减少多达 240 倍。当应用于 K562 和 HepG2 细胞时,PARM 的实测启动子活性和整体预测能力与使用全基因组 MPRA 数据获得的性能相当(图 2b)。
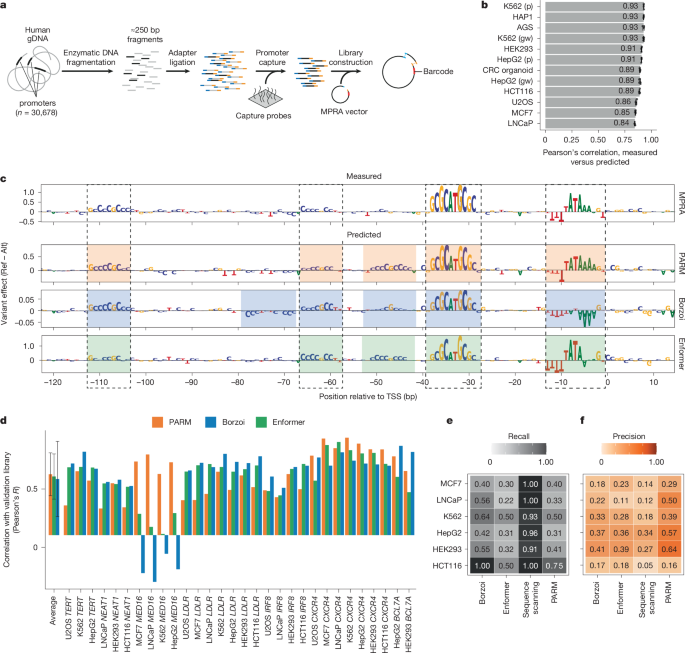
a, 启动子聚焦 MPRA 文库构建的示意图。人类 gDNA 被随机消化成约 300 bp。将片段与适配器连接,捕获启动子重叠片段并克隆到无启动子的条形码载体中,以便在转染到细胞后测量每个单独片段的自主启动子活性。b, 对于所有文库,九种细胞系和一种结直肠癌 (CRC) 类器官培养物,在 TSS 平均启动子活性(按重叠片段对每个启动子取平均值)上,实测活性与 PARM 预测活性(五种建模折叠中平均值 ± 标准差)的皮尔逊相关系数。‘gw’ 表示来自全基因组 MPRA 的数据;‘p’(以及所有其他)来自启动子聚焦 MPRA。使用模型的五个不同折叠进行预测。误差条表示这五个不同预测的标准差。c, MCF7 细胞中CXCR4启动子跨越的单核苷酸取代对活性的影响,由 MPRA、PARM、Borzoi30 和 Enformer10 如所示进行测量或预测。Alt,突变等位基因;Ref,参考基因组等位基因。d, 在七个不同细胞系中测试的七个不同启动子的单核苷酸取代效应,由 MPRA 与 PARM、Enformer 和 Borzoi 预测的相关性。对于 AGS、U2OS、HCT116 和 LNCaP 细胞,Enformer 和 Borzoi 预测不可用;因此,我们使用了来自相似细胞类型的预测(方法)。数据为平均值 ± 标准差。e,f, 由所指示的模型和通过使用 FIMO36 进行的序列扫描,识别 RSs(通过饱和突变 MPRA 检测到)的召回率 (e) 和精确率 (f)。
🚀 想要体验更好更全面的AI调用?
欢迎使用青云聚合API,约为官网价格的十分之一,支持300+全球最新模型,以及全球各种生图生视频模型,无需翻墙高速稳定,文档丰富,小白也可以简单操作。
评论区