


📢 转载信息
原文链接:https://www.nature.com/articles/s41586-026-10141-2
原文作者:Borna Novak, Jeffrey M. Lotthammer, Ryan J. Emenecker, Alex S. Holehouse
摘要
固有无序蛋白和区域(统称为IDRs)存在于所有生命王国中,并在几乎所有的真核细胞过程中发挥着关键作用1。IDRs以广泛的结构不同的构象集合形式存在。这种结构可塑性促进了多样化的分子识别和功能2,3,4。在此,我们将物理力场的进步与多模态生成式深度学习的力量相结合,开发出STARLING,一个用于快速生成准确IDR集合和从序列生成集合感知表征的框架。STARLING支持跨离子强度的环境条件,并展示了生成模型在超出其训练域进行插值能力的初步概念验证。此外,我们利用贝叶斯最大熵重加权方案,实现了在实验约束下进行集合精修。除了集合表征,STARLING序列表征可以有多种用途。我们展示了两个例子:第一,STARLING使我们能够进行基于集合的“生物物理相似体”搜索。第二,我们证明了如何利用这些潜在表征来加速从每周或每小时每个候选到秒级的、集合优先的序列设计,从而实现文库规模的设计。总之,STARLING通过新兴的生物物理特性这一视角,极大地降低了对IDR功能进行计算探究的障碍,与生物信息学蛋白序列分析形成了互补。我们根据现有实验数据评估了STARLING的准确性,并提供了一系列小插曲,说明STARLING如何能够为IDR功能快速生成假设并辅助解释实验数据。
主要内容
IDRs是结构异质的蛋白区域,估计约占真核生物蛋白质组的30%1。尽管IDRs缺乏固定的结构,但它们在转录、翻译和细胞信号传导等基本细胞过程中起着关键作用1。由于其广泛的结构异质性,IDRs必须由一个构象集合来描述:一个大型的、结构不同且不断变化的构象集合1。然而,尽管IDRs不能表示为单个3D结构,但它们确实具有序列编码的构象偏倚,并且集合在IDR功能中可能起着至关重要的作用,并且可能在疾病中受到扰动2,3,4。正如结构生物学在理解折叠结构域功能的分子基础方面发挥了重要作用一样,人们也日益认识到,对IDR集合进行表征对于理解IDR功能可能很重要5,6。
各种实验技术已被应用于探究序列–集合关系7,8,9。虽然这些技术报告了IDR集合的特定方面,但它们未能提供对构象分布的整体描述(即,蛋白质中所有残基在许多不同构象中的3D坐标,此处称为“完整结构集合”)。要实现这一点,将计算模型与实验数据相结合已被证明是一条有效的途径10,11,12,13。
从概念上讲,计算模型和实验可以以两种不同的方式结合起来。一种方法涉及使用基于物理的模型,并根据实验可观测值进行重加权或偏置14,15。另一种方法涉及使用实验数据来参数化可转移力场,这些力场原则上不需要额外的重加权16。尽管这两种方法在提供对IDR行为的洞察方面都取得了成功,但它们需要深厚的技术专长才能确保得出可靠的结论,而且它们也可能计算成本高昂。
虽然最近在粗粒度模拟17,18,19方面的进展提供了一种更快的替代方案(上述两种模式都可应用于此),但即使是粗粒度模拟,也可能需要数小时才能获得足够的采样,并且仍然需要相对较高的技术专长来设置、运行和分析。最近基于粗粒度模拟训练的深度学习预测器使得蛋白质组尺度的集合平均值预测成为可能,但它们仅限于训练预测器的特定可观测值(例如,回旋半径(Rg)或端到端距离半径(Re))20,21。
深度学习方法革新了蛋白质结构预测,通过大规模的进化信息促进了主要的进步,极大地降低了探索序列-结构关系的大门22(图 1a)。然而,这些方法不太适合研究IDRs23,原因在于无序蛋白中基于比对的保守性降低,合适的实验训练数据稀缺,以及优化目标不当(为序列预测单个最佳结构,而IDRs应由大型、构象异质的集合来描述;图 1b)。简而言之,尽管我们现在拥有用于准确预测折叠蛋白质3D结构的易用工具,但缺乏用于快速准确预测IDR集合的等效工具。
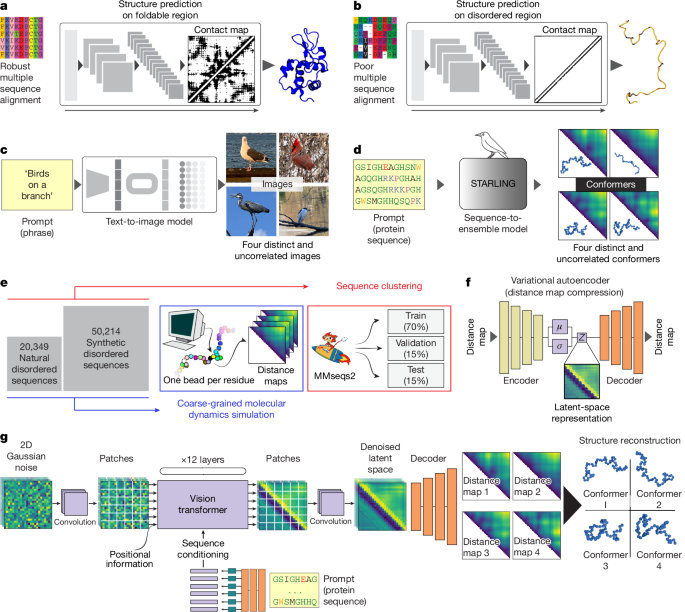
a, 深度学习彻底改变了蛋白质结构预测,大规模的进化信息促进了主要进展。b, 适用于折叠结构域的结构预测方法不太适合预测IDRs的行为。这些局限性源于缺乏天然态结构,以及进化信息在IDRs的多序列比对(MSA)中通常捕获不佳。c, 生成式文本到图像模型能够根据单个输入提示创建许多独特且独立的图像。d, IDR集合生成与文本到图像生成有许多相似之处;我们需要许多独立、不相关的构象,所有这些都与输入提示(氨基酸序列)一致。e, STARLING在约50,000个氨基酸序列上以150 mM离子强度进行了训练,在约20 mM和300 mM离子强度下对约14,000个序列进行了训练。20 mM和300 mM离子强度下的序列是150 mM离子强度下模拟序列的一个子集。对于每个序列,使用粗粒度分子动力学模拟生成了数百个不同的构象,每个构象都被转换为距离图(图像)。序列被分成训练、测试和验证集。MARV卡通图根据参考文献61,Martin Steinegger重制。f, STARLING使用VAE将距离图压缩到潜在空间,从而使去噪扩散模型可以在该潜在空间中工作(“潜在扩散”)。g, STARLING模型的整体架构结合了一个潜在空间概率去噪扩散模型和一个视觉Transformer架构,该架构同时使用卷积和Transformer块。潜在空间图通过VAE解码器解码为实空间。最后,可以通过并行化的多维标度方法将距离图重建为3D坐标。
在此,我们通过开发一种快速准确的方法,直接从氨基酸序列预测完整的粗粒度无序蛋白结构集合,来应对这些挑战。我们的方法利用了生成模型的进步,这是一种能够创建新的和原创数据的深度学习技术。然而,开发生成模型存在一个关键挑战:需要大型训练数据集。为解决此问题,我们进行了大规模的粗粒度模拟,生成了跨越数万个天然和合成IDR的完整结构集合。由此产生的方法——STARLING——使我们能够在几秒钟内直接从序列生成结构集合。开发STARLING的一个主要目标是避免硬件壁垒。尽管STARLING在GPU上速度很快(每秒约35个构象),但它仍然可以在Intel/AMD CPU上在几分钟内生成集合,在Apple CPU上在几秒钟内生成。
STARLING生成的集合与实验数据表现出良好的一致性,有助于对未表征的IDR进行从头探索或协助对实验数据的生物物理解释。此外,我们在此展示了STARLING可用于(1)研究无序蛋白的序列–集合关系;(2)探索二元无序蛋白复合物的结合态构象集合;以及(3)为IDR表征、搜索和设计提供构象感知的潜在表征。总之,我们认为STARLING的易用性和速度使其成为民主化大规模探索IDR序列–集合关系能力的强大工具。
生成式人工智能(AI)在文本到图像生成方面具有变革性24,25。在文本到图像生成式AI中,通过将提示(描述所需图像的简短短语)传递给预训练的深度学习模型来生成图像。然后,模型生成与提示一致的图像,这个过程称为推理。能够进行推理的深度学习模型必须首先经过训练。对于现代文本到图像生成式AI中使用的模型,训练不仅仅是记忆,还包括学习提示与相关图像特征之间的关系。因此,一旦模型经过训练,如果重复使用相同的提示多次,它将生成许多独立于训练中观察到的任何单个实例的图像。值得注意的是,尽管彼此不同,但每张生成的图像都应与提示一致(图 1c)。
将单个文本提示映射到一组不同图像——每张图像都与输入提示一致——正是我们希望为IDR集合生成解决的问题。在IDR集合生成中,我们希望采用文本提示(氨基酸序列)并生成与该提示一致的一组许多不同且不相关的IDR构象(图 1d)。此外,我们希望这个生成过程快速(几秒钟)并且能够在商用硬件(笔记本电脑和台式机)上进行。为实现此目标,我们将一个变分自编码器(VAE)与一个离散时间去噪扩散概率模型(DDPM)相结合,创建了一个潜在扩散模型25。由此产生的方法(STARLING)实现了对IDR粗粒度构象集合的准确快速预测。
STARLING在约50,000个独特的合理设计和天然存在的IDR序列(10–384个残基)上进行了训练,这些序列在150 mM离子强度下进行了模拟(补充表 1)。其中,约14,000个序列还在20 mM和300 mM离子强度下进行了模拟(图 1e,扩展数据图 1a,b和补充表 2)。合理设计序列是使用GOOSE26设计的(见方法;扩展数据图 1c–f)。尽管天然存在的IDR提供了聚焦于最相关序列流形的训练数据,但纳入大量合理设计序列使我们能够构建系统地贯穿序列空间的训练数据,产生一个全面、均衡的训练集,捕获序列组成和模式的极端情况。
深度学习模型中一个必要的决策是定义训练数据的范围。我们将重点放在长度最长为384个氨基酸的序列上,原因有几点。首先,常见模式生物中近95%的天然IDR长度小于384个氨基酸(扩展数据图 1h,i)。其次,该长度使得可以在合理的时间范围内完成充分采样的粗粒度模拟。第三,模型的底层架构要求一个固定的上限(见方法)。最后,对长度超过约350个残基的无序蛋白序列的实验表征很缺乏,使得评估模型对非常长的无序蛋白的预测相对于实验的有效性变得困难。
分子动力学模拟使用Mpipi-GG力场进行。Mpipi-GG20——原始Mpipi力场17的一个变体——是一种用于无序区域的单珠/残基粗粒度模型。运行后,长度为n的序列模拟产生的构象被转换为距离图,即n × n矩阵,其中每个元素描述特定构象中第i个和第j个残基之间的距离。这会将每个IDR构象转换为一个“图像”,从而使我们能够直接利用为条件图像生成开发的创新。
DDPM模型的两个核心限制是它们巨大的内存需求和推理过程中缓慢的生成过程。这些限制源于在复杂、高维空间(例如,384 × 384像素的图像)中执行反向去噪过程。为缓解此问题,我们开发了一个VAE,用于将每个距离图压缩到较低分辨率的潜在空间(24 × 24像素)27(图 1f)。然后可以在该潜在空间中进行去噪扩散过程(潜在扩散),从而显著减少内存需求和推理时间。鉴于此,STARLING分两个独立阶段进行训练。
我们首先训练了一个高精度的VAE,它能够将全分辨率距离图压缩到潜在空间(扩展数据图 2a)。我们的VAE使用ResNet18架构,并学习编码器(全分辨率到潜在空间)和解码器(潜在空间到全分辨率)的参数28。我们通过对源自与训练和验证集显著不同的序列的距离图进行编码和解码来评估VAE的准确性。我们在一个包含约16,000次模拟(总共近250万张距离图)的保留测试集上评估了我们的模型。模型实现了1.16 Å的均方根重建误差(RMSE)(扩展数据图 2b)。此外,我们的模型准确地重建了键长,均方根重建误差为0.18 Å(扩展数据图 2c),这对于模拟蛋白质构象至关重要。
在训练的第二阶段,我们开发了一个DDPM(图 1d 和扩展数据图 2d–f)。简而言之,该模型经过训练,以学习将随机噪声映射到单个潜在空间构象距离图的参数,并以相关的氨基酸序列和离子强度为条件(见方法)。训练数据来自近78,000次模拟中近1200万张距离图。每张图首先被压缩到潜在空间,然后使用固定的前向扩散过程向每张潜在空间距离图添加噪声。然后训练底层的视觉Transformer模型以学习反转该前向扩散过程的参数,并以输入氨基酸序列和离子强度为条件(图 1g)。最终,通过并行运行多次推理轮次,生成许多独立的距离图,从而实现集合生成。
尽管训练需要VAE编码器生成潜在空间距离图,但在模型训练完成后,仅需要VAE解码器进行推理。最终,完全训练好的模型(STARLING)结合了两种不同的模型(VAE解码器和DDPM),共同实现快速的集合预测。使用默认设置,STARLING可以在GPU(Nvidia A4000)上大约12秒内生成400个独立的IDR构象,在Macbook Pro M3 CPU上需要20秒,在Intel CPU(Intel(R) Xeon(R) Silver 4210 R CPU @ 2.40 GHz)上大约需要6分钟(扩展数据图 3a,b)。预测运行时间和内存大致与序列长度无关;50个氨基酸IDR的400个构象花费了大约10秒,而350个残基IDR的400个构象花费了大约14秒,并且预测时间没有受到低内存GPU的阻碍(扩展数据图 3c–e)。
我们首先检查了STARLING得出的集合平均全局尺寸是否与Mpipi-GG模拟在未见序列上的结果一致。在所有情况下,STARLING集合由800个构象组成。使用大约10,000个序列的保留测试集,我们在150 mM离子强度下进行了Mpipi-GG模拟并计算了Rg和Re。我们STARLING得出的Rg(RMSE = 0.85 Å, R2 = 0.996)和Re(RMSE = 3.48 Å, R2 = 0.989)值与模拟结果非常吻合(图 2a,b)。

a, STARLING得出的平均Rg值与来自约10,000个未见序列的Mpipi-GG(模拟)值的比较。b, STARLING得出的平均Re值与来自约10,000个未见序列的Mpipi-GG(模拟)值的比较。c, STARLING得出的Rg值与来自约3,000个未见序列、离子强度为20 mM时的Mpipi-GG(模拟)值的比较。d, STARLING得出的Rg值与来自约3,000个未见序列、离子强度为300 mM时的Mpipi-GG(模拟)值的比较。e, STARLING集合可用于计算可观测值的分布。分布相似性可通过Hellinger距离(H)量化,其中H=0为完美重叠,H=1为无重叠。f, STARLING(蓝条)和Mpipi-GG(橙线)得到的Rg分布的重叠情况,并显示了实验值作为参考。STARLING和Mpipi-GG分布之间的重叠通过H值和KS统计量(D)进行量化,以评估分布之间的差异和差异的影响程度。KS统计量0.02意味着两个分布的CDF最多相差2%。垂直虚线表示STARLING得出的平均值。g, 3D构象(左)可以表示为距离图(右),其中每个像素定义了一对残基之间的距离(rij)。h, 构象集合(左)可以表示为距离图集合(右)。i, STARLING集合中的残基内距离分布可与等效的Mpipi-GG模拟分布进行比较。j, 跨不同示例集合的所有可能的残基间距离可使用H进行比较以量化分布的重叠情况(顶部)。还显示了残基内H值的直方图(底部)。k, 跨不同示例集合的成对残基间距离比较显示出极好的重叠。
由于STARLING也在20 mM和300 mM离子强度下的Mpipi-GG模拟上进行了训练,我们验证了STARLING得出的集合平均全局尺寸与模拟结果匹配。使用大约3,000个未见序列的保留测试集,我们在20 mM和300 mM离子强度下进行了Mpipi-GG模拟。我们的STARLING得出的Rg(图 2c,d;对于20 mM和300 mM,RMSE分别为0.98 Å和1.12 Å, R2分别为0.995和0.992)和Re(扩展数据图 4a,b;对于20 mM和300 mM,RMSE分别为4.17 Å和4.53 Å, R2分别为0.986和0.978)再次与模拟结果高度一致。
模型准确性取决于集合大小和去噪步骤之间的平衡。通过将STARLING结果与长时间尺度的Mpipi-GG模拟进行比较,我们发现Rg误差在超过30个去噪步骤和400个构象后趋于稳定(扩展数据图 4c–i)。我们将这些视为默认参数,尽管对于其他排序参数可能需要更大的集合。
由于Re和Rg与序列长度高度相关,我们接下来评估了模型在一组长度匹配序列29上的准确性。这种方法使我们能够确定模型是否有效地捕获了序列化学对IDR全局尺寸的影响,还是仅仅学会了将序列长度与这些尺寸相关联。使用保留测试集中具有不同序列化学特性的所有100残基、200残基和300残基序列,我们发现在Rg(扩展数据图 4j;对于100残基、200残基和300残基长度,RMSE分别为0.78 Å, 1.18 Å和1.20 Å, R2分别为0.984, 0.990和0.995)和Re(扩展数据图 4k;对于100残基、200残基和300残基长度,RMSE分别为3.38 Å, 4.22 Å和5.65 Å, R2分别为0.965, 0.984和0.986)的各个范围内都存在极好的一致性。这些结果使我们确信STARLING学习到的是真正的序列到集合规则,而不仅仅是聚合物缩放理论。
除了集合平均可观测值,完整的结构集合还使我们能够计算任何感兴趣的可观测值的分布(图 2e)。我们着手评估STARLING得出的全局尺寸分布与模拟得到的分布匹配的程度。我们使用Hellinger距离(H)作为相似性度量来比较分布。H的范围从0到1,其中零表示分布相同,而1表示它们完全不相交(图 2e)。STARLING得出的Rg分布与模拟分布显示出极好的一致性(图 2f)。这种重叠通过较低的H值量化,证实了两个分布的高度相似性。
最后,为了量化集合保真度,我们评估了使用STARLING或Mpipi-GG模拟生成的集合之间所有残基间距离分布的差异。每个3D构象可以表示为单个距离图(图 2g),而构象集合可以表示为距离图集合(图 2h)。因此,可以将STARLING集合中一对残基的每个残基间距离分布与来自Mpipi-GG的相应分布进行比较(图 2i)。对于一个100残基的IDR,这产生了4,900个唯一的分布比较。我们可以使用H量化分布之间的重叠,并使用热图可视化每对(图 2j,顶部)。此外,我们可以使用H的直方图来提供对集合一致性的总体评估(图 2j,底部)。总而言之,我们发现所有研究的距离分布都存在极好的一致性(图 2k)。总之,所有证据都支持STARLING可以直接从序列预测无序蛋白的构象集合。
在确定STARLING可以重现模拟集合之后,我们接下来研究了与实验数据的一致性(图 3a)。我们使用了一组先前整理的133个序列,这些序列已收集了高质量的小角X射线散射(SAXS)数据,并计算了平均Rg值。我们发现一致性极好…… [内容被截断]
🚀 想要体验更好更全面的AI调用?
欢迎使用青云聚合API,约为官网价格的十分之一,支持300+全球最新模型,以及全球各种生图生视频模型,无需翻墙高速稳定,文档丰富,小白也可以简单操作。
评论区