


📢 转载信息
原文链接:http://bair.berkeley.edu/blog/2025/04/08/plaid/
原文作者:BAIR (Berkeley Artificial Intelligence Research)
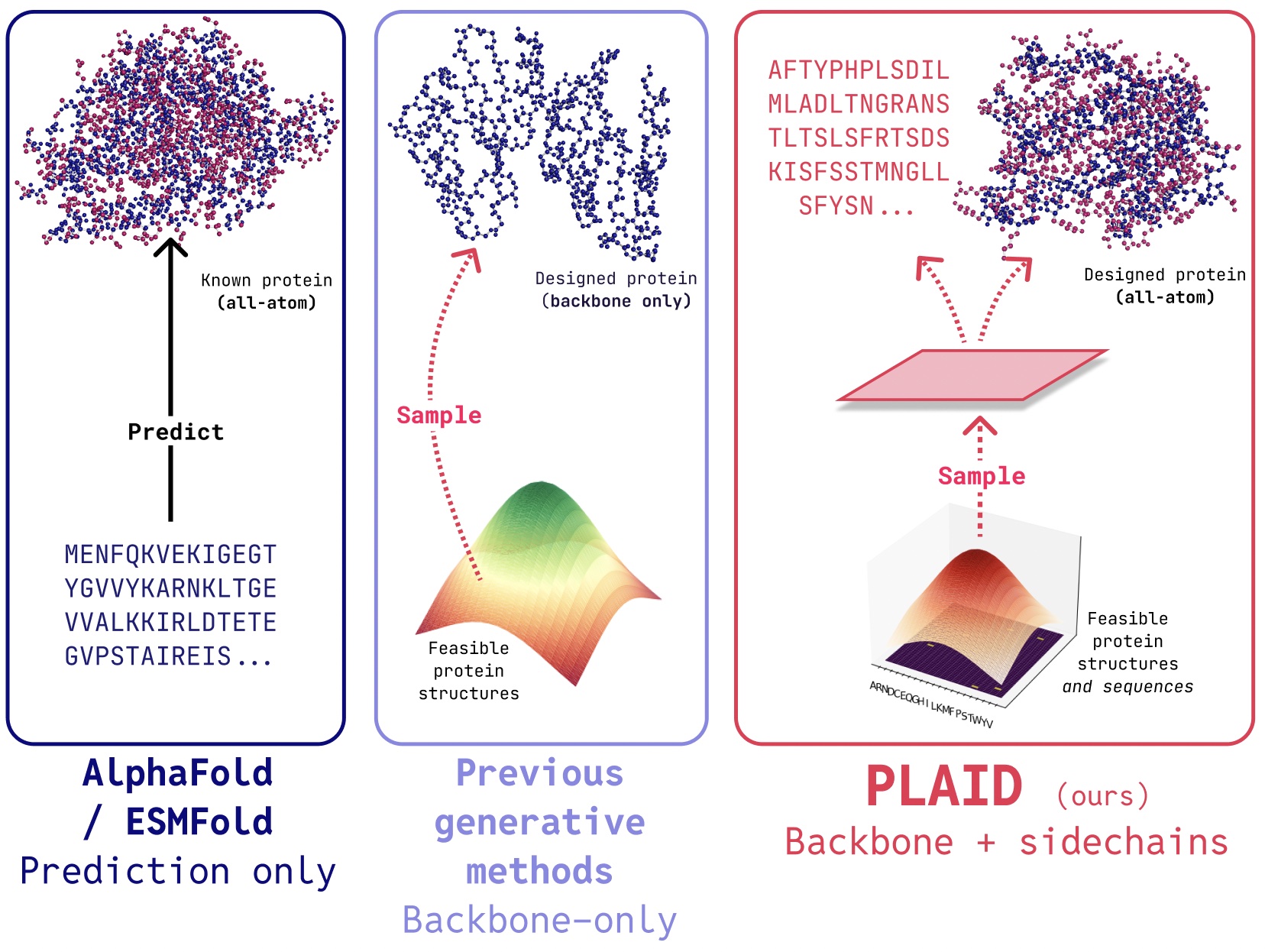
PLAID 是一种多模态生成模型,它通过学习蛋白质折叠模型的潜在空间,可以同时生成蛋白质的1D序列和3D结构。
2024年诺贝尔奖授予AlphaFold2,标志着人工智能在生物学领域的作用得到了重要认可。蛋白质折叠之后的下一步是什么?
在PLAID中,我们开发了一种方法,可以学习从蛋白质折叠模型的潜在空间中采样,从而生成新的蛋白质。它可以接受组合性的功能和生物体提示,并且可以在序列数据库上进行训练,这些数据库比结构数据库大2到4个数量级。与许多现有的蛋白质结构生成模型不同,PLAID解决了多模态联合生成问题:同时生成离散的序列和连续的全原子结构坐标。
从结构预测到真实世界的药物设计
尽管最近的研究表明扩散模型在生成蛋白质方面显示出潜力,但现有模型仍然存在一些限制,使其不适用于现实世界的应用,例如:
- 全原子生成:许多现有的生成模型只产生骨架原子。要生成全原子结构并放置侧链原子,我们需要知道序列。这创建了一个需要同时生成离散和连续模态的多模态生成问题。
- 生物体特异性:用于人体使用的蛋白质生物制剂需要被人性化,以避免被人体免疫系统破坏。
- 控制规范:药物发现并将其交给患者是一个复杂的过程。我们如何指定这些复杂的约束条件?例如,即使解决了生物学问题,您也可能决定药片比小瓶更容易运输,从而对溶解性增加了一个新的约束。
生成“有用”的蛋白质
简单地生成蛋白质不如控制生成以获得有用的蛋白质那么有价值。这样的界面可能是什么样子?

为了获得灵感,让我们考虑如何通过组合性文本提示来控制图像生成(示例来自Liu et al., 2022)。
在PLAID中,我们为控制规范镜像了这种界面。最终目标是通过文本界面完全控制生成,但在这里我们考虑了两个轴的组合约束作为概念验证:功能和生物体:
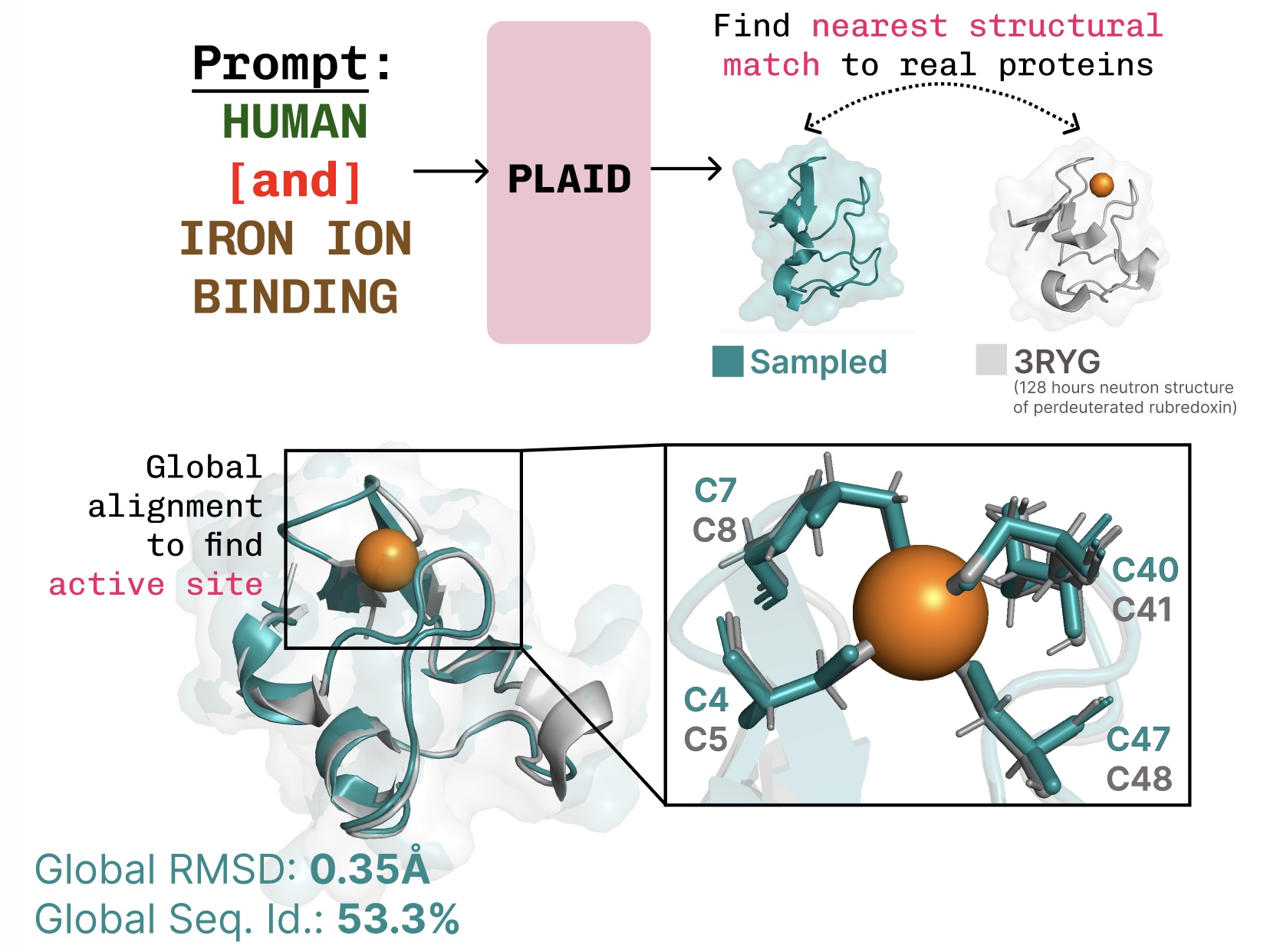
学习功能-结构-序列的联系。 PLAID学习了通常在金属蛋白中发现的四面体半胱氨酸-Fe2+/Fe3+配位模式,同时保持高序列水平的多样性。
使用仅序列的训练数据进行训练
PLAID模型的另一个重要方面是,我们只需要序列即可训练生成模型!生成模型学习由其训练数据定义的分布,并且序列数据库比结构数据库大得多,因为获得序列比获得实验结构便宜得多。
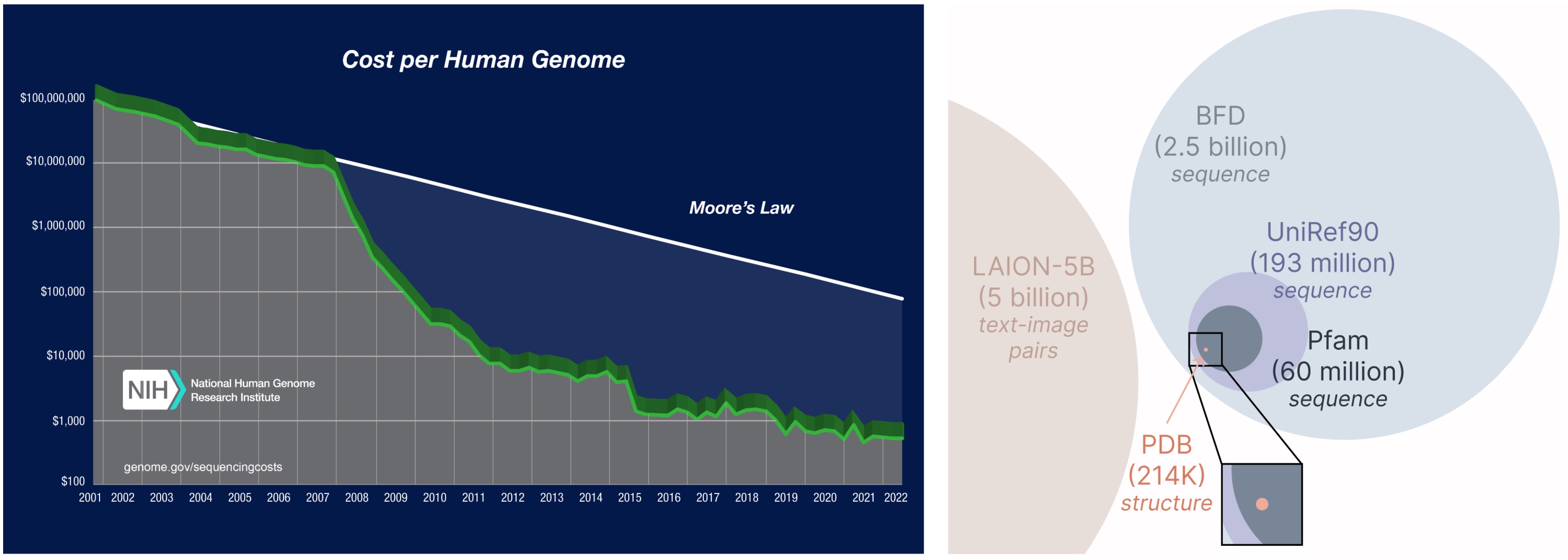
从更大、更广泛的数据库中学习。 获取蛋白质序列的成本远低于实验表征结构,并且序列数据库比结构数据库大2到4个数量级。
它是如何工作的?
我们能够仅使用序列数据训练生成模型来生成结构的原因是,我们学习了蛋白质折叠模型的潜在空间上的扩散模型。然后在推理过程中,从有效蛋白质的潜在空间中采样后,我们可以从蛋白质折叠模型中采用冻结的权重来解码结构。在这里,我们使用ESMFold,它是AlphaFold2模型的后续版本,它用蛋白质语言模型取代了检索步骤。
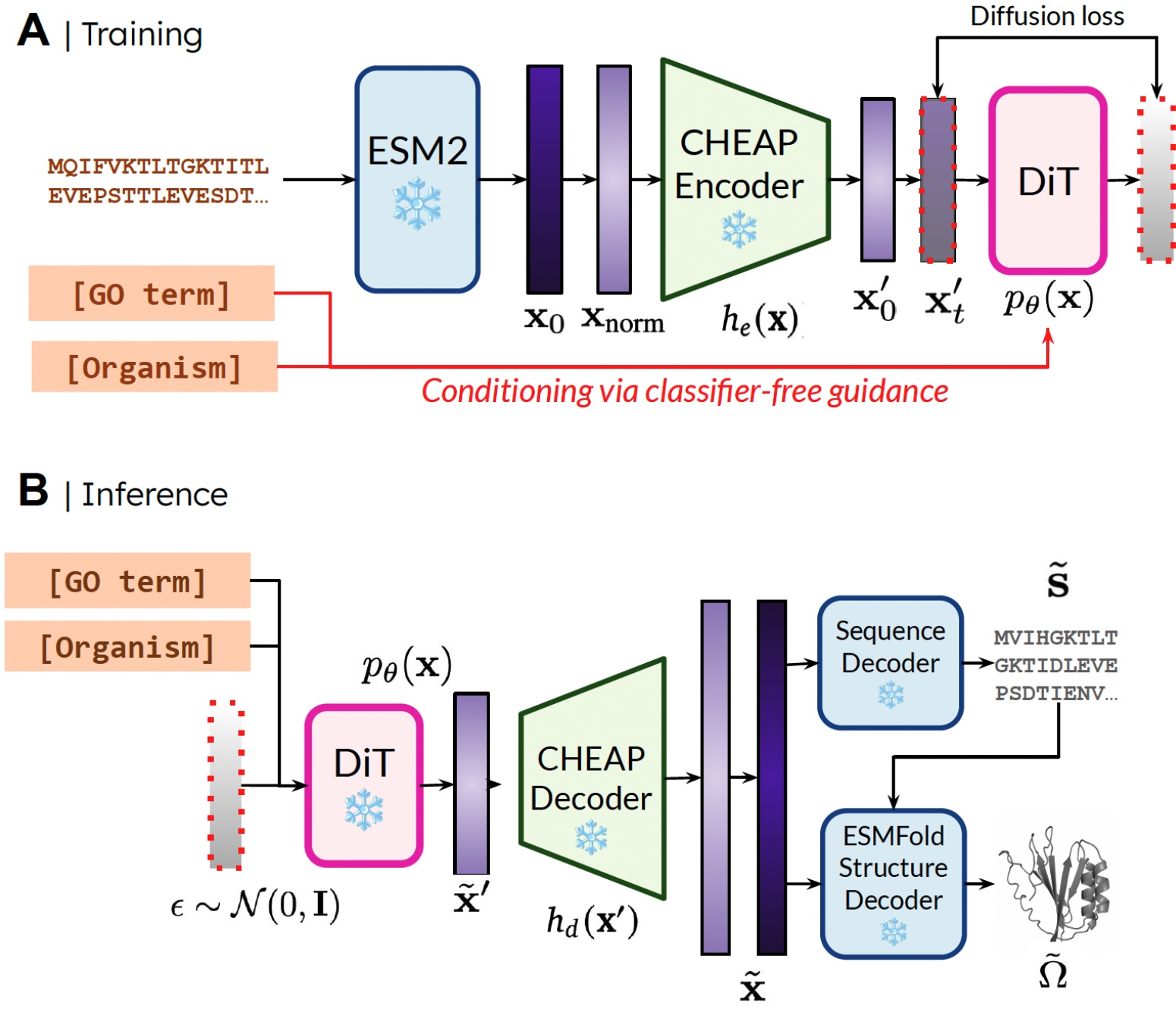
我们的方法。 在训练期间,只需要序列即可获得嵌入;在推理期间,我们可以从采样的嵌入中解码序列和结构。❄️表示冻结的权重。
通过这种方式,我们可以利用预训练蛋白质折叠模型中结构理解信息用于蛋白质设计任务。这类似于机器人技术中的视觉-语言-动作(VLA)模型如何利用在互联网规模数据上训练的视觉-语言模型(VLM)中包含的先验知识,以提供感知、推理和理解信息。
压缩蛋白质折叠模型的潜在空间
直接应用此方法的一个小难点是ESMFold的潜在空间——事实上,许多基于Transformer的模型(Transformer-based models)的潜在空间——需要大量的正则化。这个空间也非常大,因此学习这个嵌入最终会映射到高分辨率的图像合成。
为了解决这个问题,我们还提出了CHEAP (Compressed Hourglass Embedding Adaptations of Proteins,蛋白质的压缩沙漏嵌入适应),我们学习一个压缩模型来联合嵌入蛋白质序列和结构。
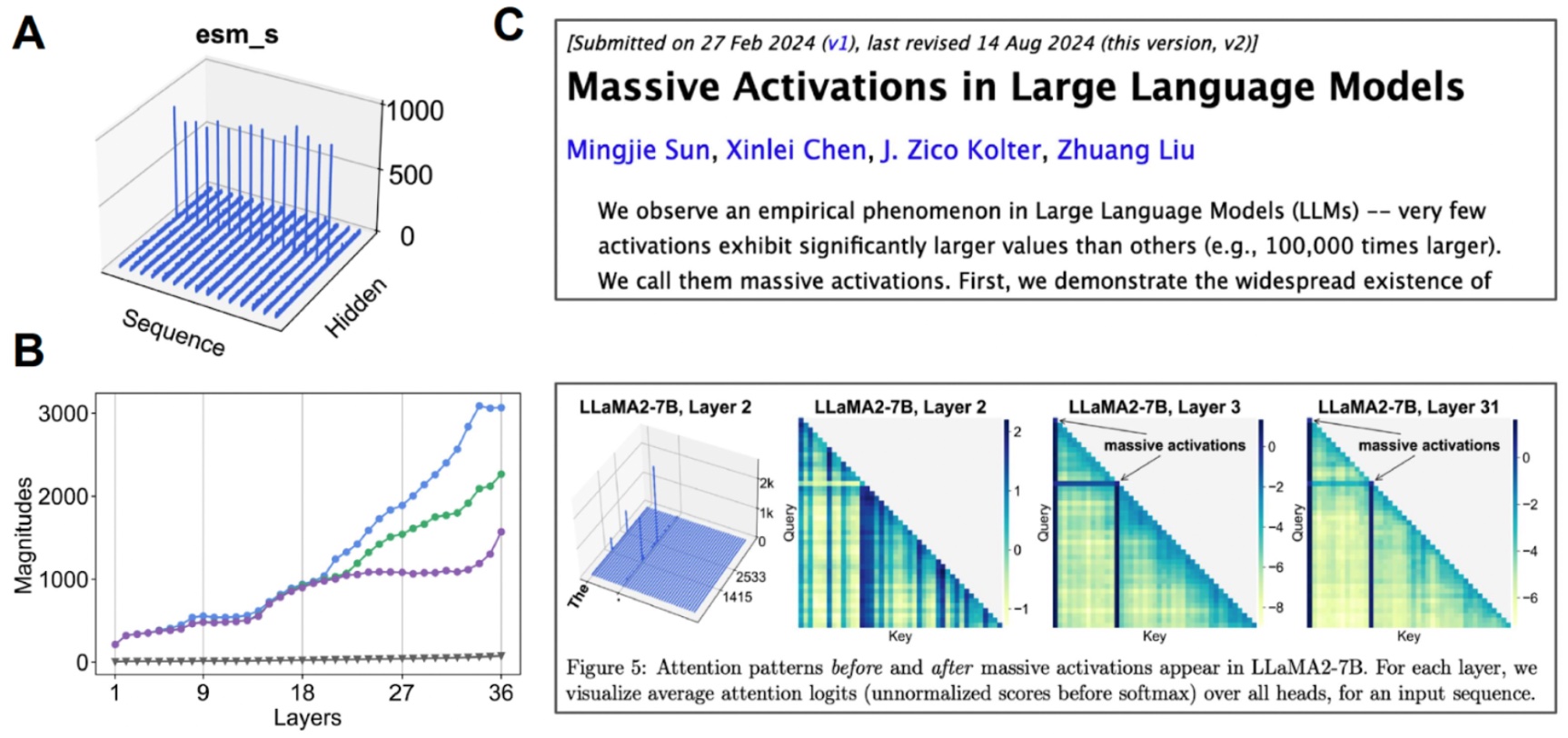
探究潜在空间。 (A) 当我们可视化每个通道的平均值时,一些通道表现出“巨大激活”。(B) 如果我们开始检查与中值(灰色)相比的前3个激活值,我们发现这种情况发生在许多层中。(C) 对于其他基于Transformer的模型,也观察到了巨大激活。
我们发现这个潜在空间实际上是高度可压缩的。通过对我们正在处理的基础模型进行一些机械可解释性研究,我们能够创建一个全原子蛋白质生成模型。
下一步是什么?
尽管在这项工作中我们研究了蛋白质序列和结构生成的案例,但我们可以将此方法改编为对任何模态执行多模态生成,只要存在从更丰富的模态到更稀疏模态的预测器即可。随着蛋白质的序列到结构预测器开始处理越来越复杂的系统(例如,AlphaFold3也能够预测与核酸和分子配体复合物中的蛋白质),可以很容易地想象使用相同的方法对更复杂的系统执行多模态生成。如果您有兴趣合作扩展我们的方法,或在湿实验室中测试我们的方法,请与我们联系!
更多链接
如果您在研究中发现我们的论文有用,请考虑使用以下PLAID和CHEAP的BibTeX:
@article{lu2024generating, title={Generating All-Atom Protein Structure from Sequence-Only Training Data}, author={Lu, Amy X and Yan, Wilson and Robinson, Sarah A and Yang, Kevin K and Gligorijevic, Vladimir and Cho, Kyunghyun and Bonneau, Richard and Abbeel, Pieter and Frey, Nathan}, journal={bioRxiv}, pages={2024--12}, year={2024}, publisher={Cold Spring Harbor Laboratory}
}@article{lu2024tokenized, title={Tokenized and Continuous Embedding Compressions of Protein Sequence and Structure}, author={Lu, Amy X and Yan, Wilson and Yang, Kevin K and Gligorijevic, Vladimir and Cho, Kyunghyun and Abbeel, Pieter and Bonneau, Richard and Frey, Nathan}, journal={bioRxiv}, pages={2024--08}, year={2024}, publisher={Cold Spring Harbor Laboratory}
}您还可以查看我们的预印本(PLAID, CHEAP)和代码库(PLAID, CHEAP)。
一些额外的蛋白质生成乐趣!
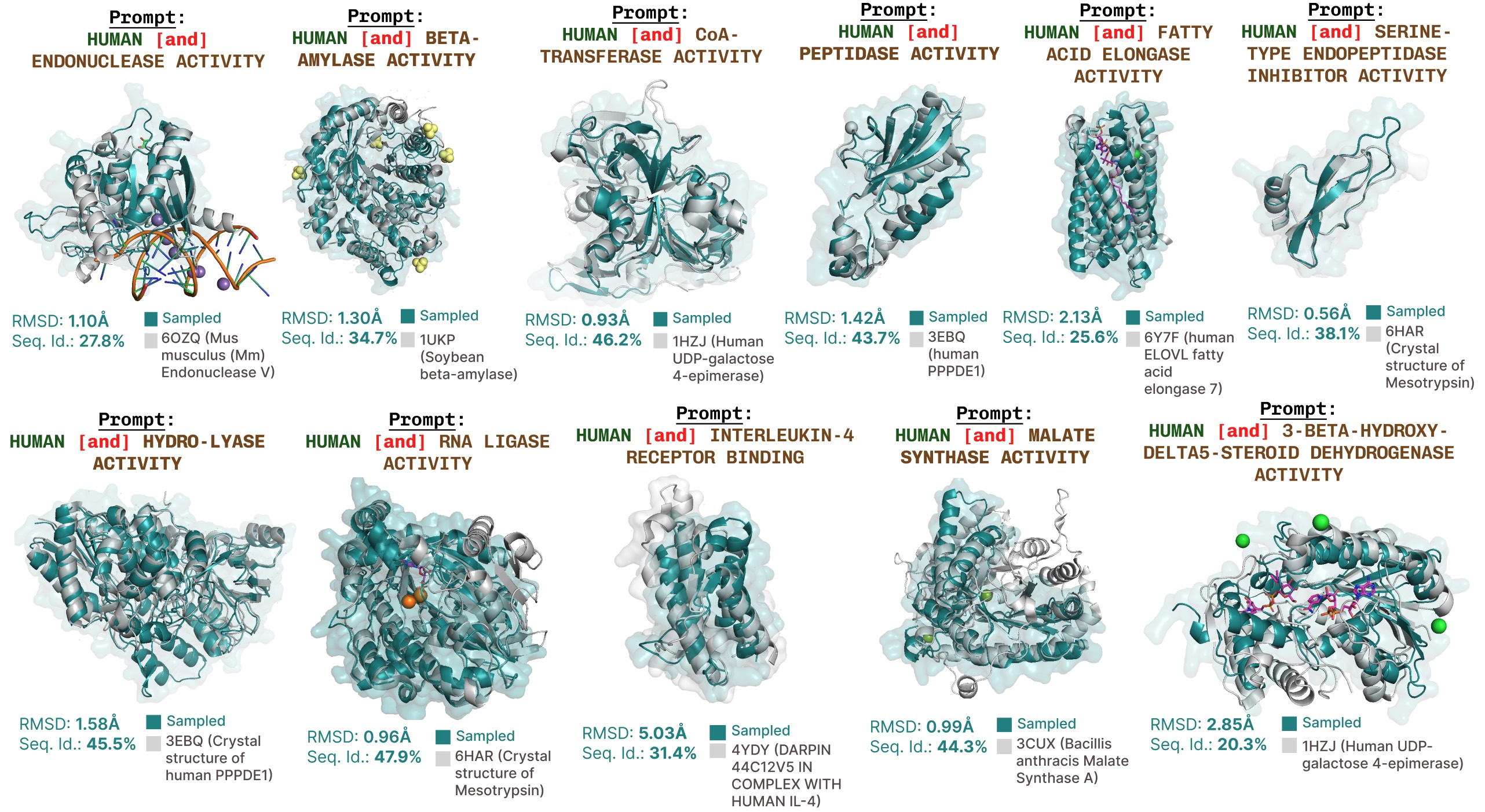
PLAID附加的功能提示生成。
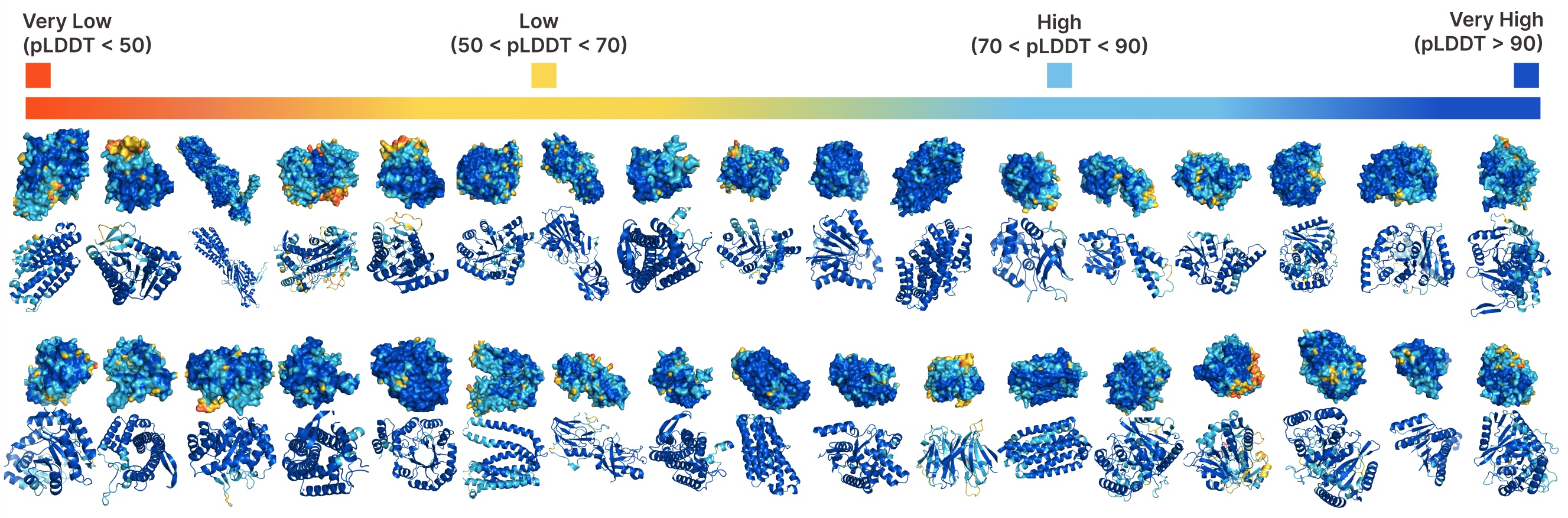
PLAID的无条件生成。
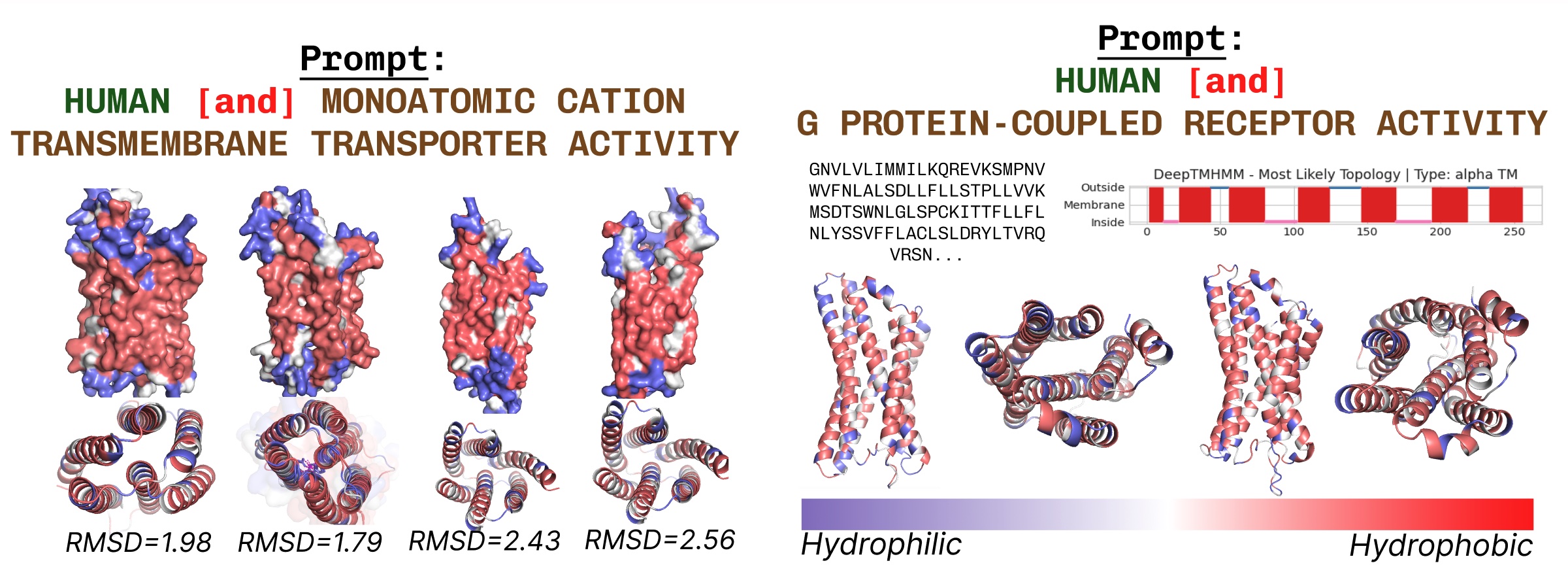
跨膜蛋白在脂肪酸层嵌入的核心处具有疏水残基。当使用跨膜蛋白关键词提示PLAID时,这些特征是一致观察到的。
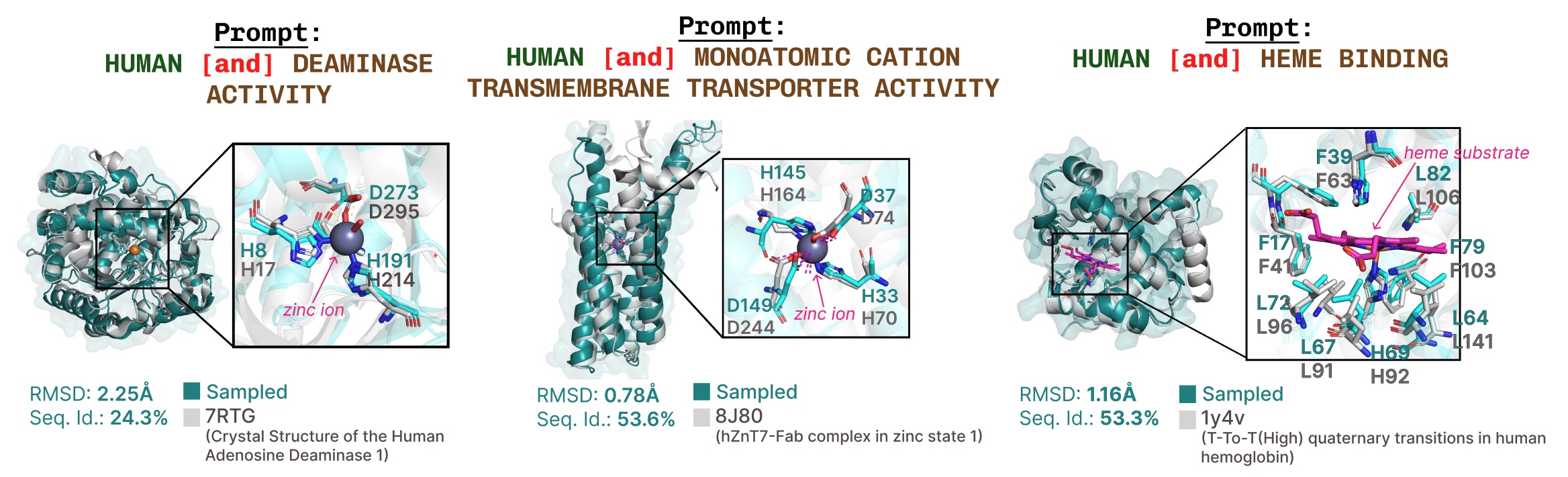
基于功能关键词提示的活性位点再现的其他示例。
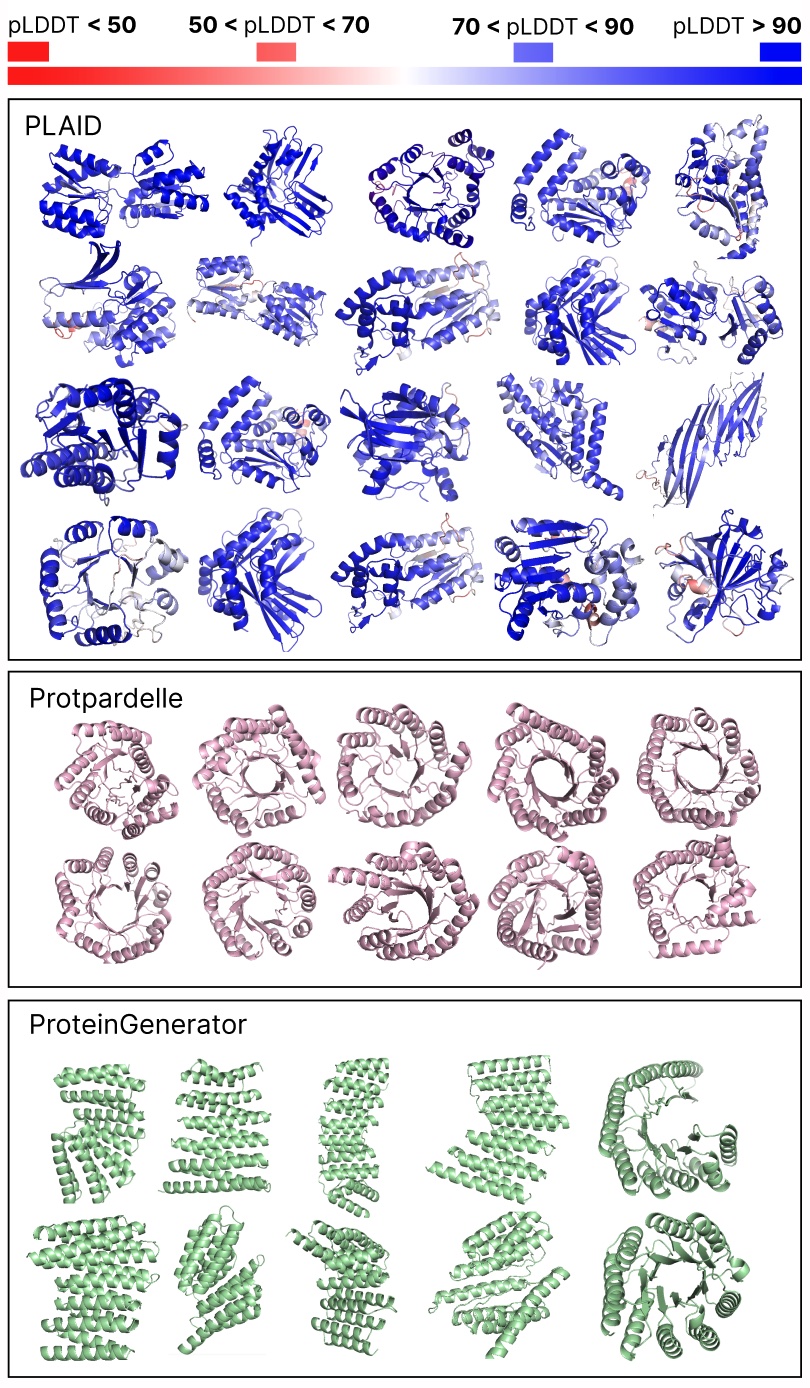
比较PLAID与全原子基线之间的样本。PLAID的样本具有更好的多样性,并捕获了对蛋白质生成模型来说更难学习的β折叠模式。
致谢
感谢Nathan Frey对本文提供的详细反馈,以及BAIR、基因泰克(Genentech)、微软研究院(Microsoft Research)和纽约大学(New York University)的合作作者:Wilson Yan, Sarah A. Robinson, Simon Kelow, Kevin K. Yang, Vladimir Gligorijevic, Kyunghyun Cho, Richard Bonneau, Pieter Abbeel, 和 Nathan C. Frey。
🚀 想要体验更好更全面的AI调用?
欢迎使用青云聚合API,约为官网价格的十分之一,支持300+全球最新模型,以及全球各种生图生视频模型,无需翻墙高速稳定,文档丰富,小白也可以简单操作。
评论区